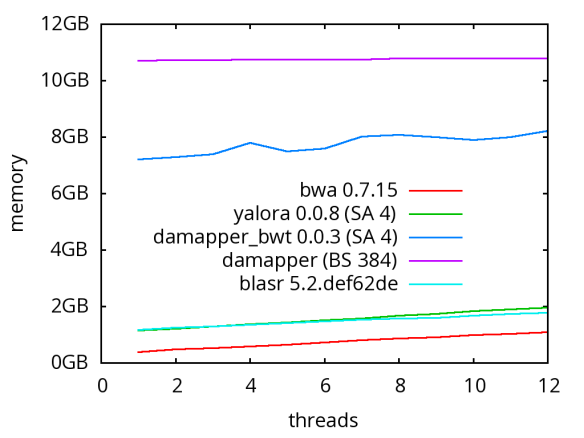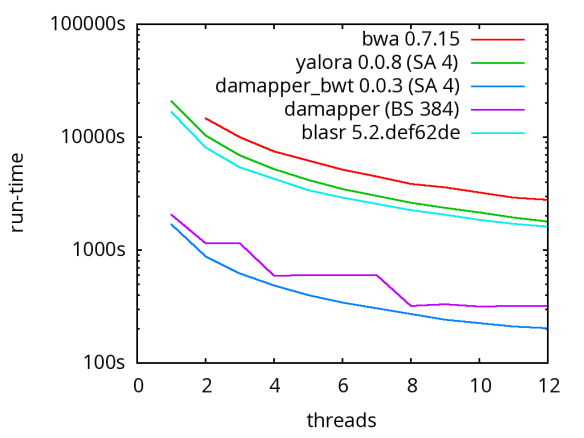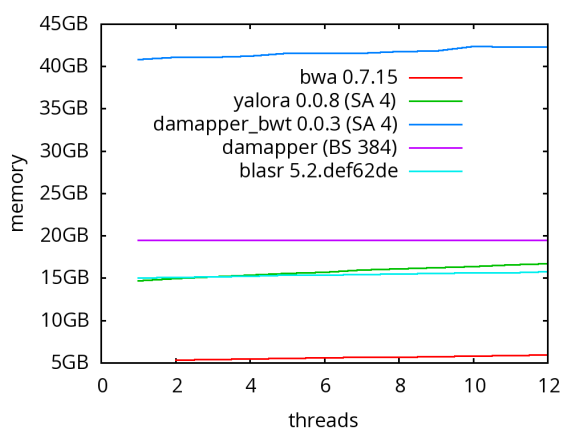
In an earlier post, I introduced the concept of intrinsic quality values — a statistic indicating the quality of every say TP=100bp interval of a read — that could be computed from the pile of local alignments for each read computed by daligner with the -t option set to TP. This idea is important because of the complex and variable error profile of PacBio long reads which I illustrated with the following hypothetical example:

Error profile of a typical long read. The average error rate is say 12% but it varies and occasionally is pure junk.
The quality values together with the pattern of alignments in a read’s pile allow one to detect these, sometimes long, low-quality segments of a read, as well as detect when the read is a chimer or contains undetected adaptamer sequence that should have been excised. These artifacts are quite frequent in PacBio data sets and apart from repeats are the major cause of poor assembly. We call the process of repairing the low-quality sequence and removing the chimers and missed adaptamers, scrubbing, and formally the goal of a scrubber is to edit a set of reads with as little loss of data as possible so that:
- every edited read is a contiguous segment of the underlying genome being sequenced, and
- every portion of a read is of reasonable quality, i.e. <20% error or Q8.
The conditions above are exactly those assumed by a string graph construction algorithm and any data that doesn’t meet these conditions introduces breaks and false branches in the string graph. For this reason I choose to scrub hard, i.e. my scrubber will over call chimers and adaptamers rather than miss them, so that condition 1 above is meet by all the data after scrubbing.
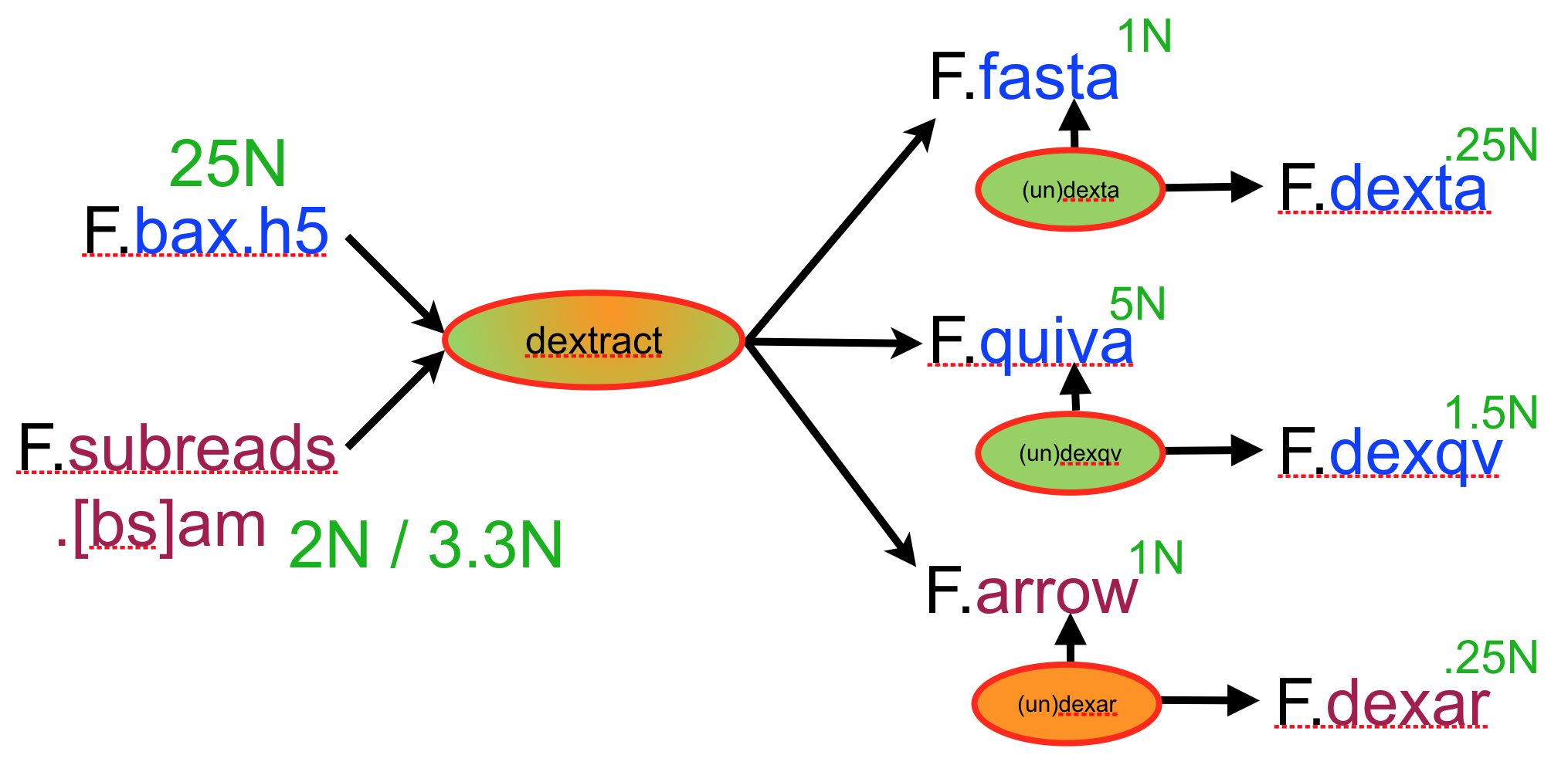
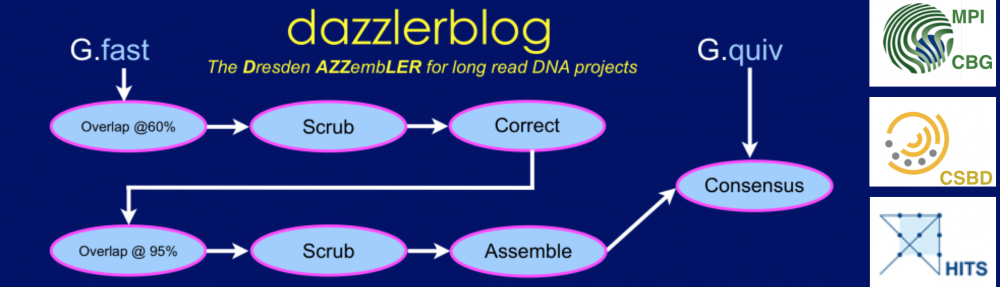
The scrubber module consists of a pipeline of 4-5 modules as opposed to a single program that performs the end-to-end task. We structured it this way so that one can run each pass in a block-parallel manner on an HPC cluster, and secondarily, so that a creative user can potentially customize or improve aspects of the overall process. The figure below, shows the pipeline:

Data flow, including tracks, of the scrubbing pipeline
Each program in the pipeline can be run on the overlap piles for a block of the underlying DB in parallel, allowing a simple HPC parallel scheme. By doing so each program per force outputs only the portion of a track for the intput block (i.e.. .qual, .trim, .patch), and these block tracks must be concatenated into a single track for the entire database with Catrack before the next step in the pipeline can be performed. Fortunately, this serial step is very fast, basically amounting to concatenating a number of files of total size O(R) where R is the number of reads in the database.
The first step in scrubbing is to compute the quality values for every read segment and record them in a qual track with DASqv as described in an earlier post. Recall that with the -v option DASqv displays a histogram of the QV’s over the dataset. Using this histogram one should select a threshold GOOD for which 80% of the QV’s are not greater than this value, and another BAD for which 5-7% of the QV’s are not less than this value. In the next two steps of the pipeline, QV’s ≤ GOOD will be considered “(definitley) good”, and QV’s ≥ BAD will be considered “(definitely) bad”. Any QV’s between the two thresholds will be considered of unknown quality. Decreasing these values, increases the amount of data that will be scrubbed away as not of sufficient quality, and increasing these thresholds retains more data but at a sacrifice in quality. The percentile guidelines above represent good settings for typical PacBio data (that is about 80% of the data actually is good and 5% of it is actually crap).
of the QV’s are not less than this value. In the next two steps of the pipeline, QV’s ≤ GOOD will be considered “(definitley) good”, and QV’s ≥ BAD will be considered “(definitely) bad”. Any QV’s between the two thresholds will be considered of unknown quality. Decreasing these values, increases the amount of data that will be scrubbed away as not of sufficient quality, and increasing these thresholds retains more data but at a sacrifice in quality. The percentile guidelines above represent good settings for typical PacBio data (that is about 80% of the data actually is good and 5% of it is actually crap).
The GOOD/BAD thresholds must be supplied as the -g and -b parameters to the next phase of the pipeline realized by DAStrim. DAStrim first uses the quality values and the supplied -g and -b thresholds to determine a sequence of high-quality (HQ) segments for the A-read of each pile. Formally, an HQ segment begins and ends with a good trace point interval interval, contains no bad intervals, and is at least 400bp long. Any intervals before the first HQ segment and after the last HQ segment, are clipped/trimmed from the beginning and end of the read, respectively. The main task of DAStrim, is to then determine what to do with the gaps between the HQ segments of a read. There are 4 cases:
- The gap is completely covered/spanned by lots of
 local alignments with other reads – then the gap is simply low quality sequence and deemed LOW-Q. This is illustrated in the example at right by the leftmost low-quality gap.
local alignments with other reads – then the gap is simply low quality sequence and deemed LOW-Q. This is illustrated in the example at right by the leftmost low-quality gap. - The gap is not spanned by local alignments with other reads, but most of the local alignments to the left (X) and right (Y) of the gap are for the same B-read and the distance between the two B-read segments in the relevant local alignments are consistent with the size of the gap – then the gap is a (very) low quality (and often long) drop out in the A-read and the gap is deemed LOW-Q.
- the gap is not spanned but the left (X) and right (Y) alignment sets have the relevant B-reads in the complement orientation with respect to each other – the gap is due to an ADAPTAMER. Typically, the gap is also very sharp with the gap between paired local alignments being very narrow as in the example below left. In the situation where one or more adatamer gaps is found for a read, all but the longest subread between two of the adaptamers is thrown away.



- the gap is not spanned and there is no correspondence between the reads in the local alignments to the left and right – the gap is due to a chimeric join in the A-read and the gap is designated CHIMER as illustrated in the example above right. Every chimeric gap splits the A-read into two new reads.
DAStrim outputs a track (or track block) called trim that for each read encodes the begin/end coordinates for each HQ segment and the classification of each intervening gap (either LOW-Q or CHIMER). With the -v option it also outputs a report describing how much data was trimmed, designated LOW-Q, etc. In the example below, one sees that the left column is the number of reads or items, and the right column is the total number of bases in those items. 3,117 reads totalling 11.6% of the bases in the data set were found to be either garbage or were totally masked by the DAMASKER module and thrown out. Of the remaining reads, 5.7% of the bases were trimmed from the beginning of reads, and 2.5% from the end of reads. 37 missed adaptamer sites were found and 117.8Kbp was trimmed away leaving only the longest sub-read between adaptamers.
Input: 22,403 (100.0%) reads 250,012,393 (100.0%) bases
Discard: 3,117 ( 13.9%) reads 29,036,136 ( 11.6%) bases
5' trim: 13,749 ( 61.4%) reads 14,137,800 ( 5.7%) bases
3' trim: 7,919 ( 35.3%) reads 6,316,157 ( 2.5%) bases
Adapter: 37 ( 0.2%) reads 117,816 ( 0.0%) bases
Gaps: 35,253 (157.4%) gaps 15,205,800 ( 6.1%) bases
Low QV: 30,903 (137.9%) gaps 8,834,200 ( 3.5%) bases
Span'd: 3,346 ( 14.9%) gaps 4,441,700 ( 1.8%) bases
Chimer: 1,004 ( 4.5%) gaps 1,929,900 ( 0.8%) bases
Clipped: 25,826 clips 49,607,909 ( 20.6%) bases
Patched: 34,249 patches 13,275,900 ( 5.3%) bases
In the adaptamer-free reads that were retained, there were 35,253 gaps between HQ segments. 30,903 were deemed LOW-Q by virtue of being spanned by alignments (Low QV) and 3,346 were deemed LOW-Q by virture of consistent alignment pairs (Span’d). The remaining 1,004 where deemed to be CHIMERs and will be split creating 1,004 new reads. The last two lines tell one that a total of 20.6% of the bases were lost altogether because of trimming, adapatmers, and chimers, and that 5.3% of the bases were in low-quality gaps that will be patched with better sequence in the ensuing scrubbing passes.
The 3rd phase of scrubbing is realized by DASpatch which selects a good B-read segment to replace each LOW-Q gap with. We call this high-quality sequence a patch. The program also reviews all the gap calls in light of the fact that the trimming of reads affects their B-read alignment intervals in the piles of other reads, occasionally implying that a gap should have been called a CHIMER and not LOW-Q. DASpatch further changes a LOW-Q gap to a CHIMER call if it cannot find what it thinks is a good B-read segment to patch the gap. The number of these calls is generally very small, often 0, and their number is reported when the -v option is set. DASpatch outputs a track (or track block) called patch that encodes the sequence of B-read segments to use for patching each low quality gap in a read.
The final phase of scrubbing is to perform the patching/editing of reads that is encoded in the trim and patch tracks produced in the last two phases, and produce a new data base of all the resulting scrubbed reads. This is accomplished by DASedit that takes the name of the original database and the name you would like for the new database as input. This phase runs sequentially and can can be quite slow due to the random access pattern of read sequences required to patch, so you might want to go do something else while it runs. But when its finished you will have a database of reads that are typically 99.99% chimer and adatamer free, and that contain almost no stretches of very low-quality base calls.
The new database does not have a .qvs or .arr component, that is it is a a sequence or S-database (see the original Dazzler DB post). Very importantly, the new database has exactly the same block divisions as the original. That is, all patched subreads in a block of the new database have been derived from reads of the same block in the original database, and only from those reads. DASedit also produces a map track that for each read encodes the original read in the source DB that was patched and the segments of that read that were high-quality (i.e. not patched). A program DASmap output this information in either an easy-to-read or an easy-to-parse format. A small example of a snippet output by DASmap is as follows:
55 -> 57(2946) [400,2946]
56 -> 58(11256) [700,1900]
57 -> 58(11256) [6600,9900] 83 [10000,11256]
58 -> 59(12282) [400,4100] 88 [4200,9400] 97 [9500,12282]
The first line indicates that read 55 in the patched database was derived from read 57 in the original database and is the segment from [400,2946] of that read. Reads 56 and 57 were both derived from read 58 in the original DB, and read 57 consists of segments [6600,9900] and [10000,11256] of read 58 with a patch between them of 83bp (but the source of the patch data is not given). The read length of each original read is given for convenience. The purpose of this map is to allow you to map back to the original reads in the final consensus phase of an assembly where one will want to use the original data along with its Quiver or Arrow data encoded in the .qvs/.arr component of the DB.
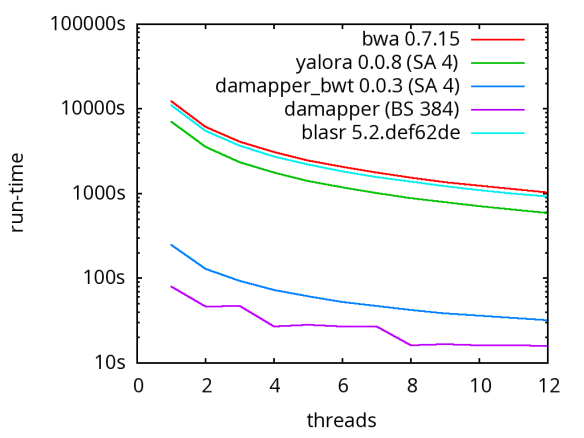
A database of scrubbed reads can be used in three ways as illustrated in the figure at the end of this paragraph. In the first use-case, a user can simply take the database of scrubbed reads and use them as input to their favorite long-read assembler (e.g. Falcon or Canu). In our experience doing so improves the assemblies produced by t hese third party systems. For example, in the plot at right, Falcon’s error corrector (EC) tends to break reads at long low quality gaps producing the red line read-length profile given a data set with the blue read-length distribution. When Falcon starts on our scrubbed reads with the grey read-length profile, (which is significantly better than the EC profile), its own error correct leaves these longer scrubbed reads intact. Longer reads into the string graph phase of Falcon’s assembler implies a more coherent assembly, i.e. longer contigs.
hese third party systems. For example, in the plot at right, Falcon’s error corrector (EC) tends to break reads at long low quality gaps producing the red line read-length profile given a data set with the blue read-length distribution. When Falcon starts on our scrubbed reads with the grey read-length profile, (which is significantly better than the EC profile), its own error correct leaves these longer scrubbed reads intact. Longer reads into the string graph phase of Falcon’s assembler implies a more coherent assembly, i.e. longer contigs. The second use-case, is to rerun the daligner on this set of reads to produce .las files anew over the scrubbed reads. The down side is that this takes considerable compute time. However, because the reads are scrubbed, one should expect that reads properly overlap and a purely local alignments can now be ignored as repeat-induced. Giving the result set of overlaps to a string graph algorithm for assembly should result in better assembly as many repeat-induced correspondences have been removed.
The second use-case, is to rerun the daligner on this set of reads to produce .las files anew over the scrubbed reads. The down side is that this takes considerable compute time. However, because the reads are scrubbed, one should expect that reads properly overlap and a purely local alignments can now be ignored as repeat-induced. Giving the result set of overlaps to a string graph algorithm for assembly should result in better assembly as many repeat-induced correspondences have been removed.
The final third use-case, is rather than re-run daligner, to take advantage of the fact that the piles computed the first time already contain all the pairs of reads that align, and so in fact all that needs to be done is to “re-align” an local alignment found between original reads in terms of the derived reads. This includes extending the local alignment when the introduction of a patch allows it to now extend through a formerly low-quality region. To this end I produced a program DASrealign that takes a pair of blocks of the scrubbed DB and the original .las file for the block pair (in the original DB), and produces the realigned overlap piles in a .las file for the scrubbed DB blocks. Not carefully that this requires (1) that you have kept the block pair .las files produced by the first phase of the HPC.daligner process and not removed them, and (2) that after realigning every block pair, you must then merge these to produce the overlap blocks for the scrubbed DB. This process is roughly 40x faster than running daligner from scratch. The trade off is that some alignments might be missed that would otherwise be found if daligner were rerun on the scrubbed reads, and the trace point encoding of a patched alignment is no longer uniform in the A-read and so one could not scrub the patched reads a second time with the piles so produced. On the otherhand, one can take just the overlaps that are produced this way and perform a string graph assembly.
The pipeline above careful identifies and removes read artifacts such as chimers and adapatamers, and repairs high error-rate read segments, providing a set of reads that will assemble well (without correction). However, ultimately we believe good assembly will further require correction in order to carefully separate haplotypes and low copy number repeats prior to the string graph phase. Therefore, our ultimate ongoing development plan is to replace DASedit with a program that produces error corrected A-reads based on the analysis of DASqv-DAStrim-DASpatch and to then realign (at much higher stringency / lower error rate) the resulting reads. We expect this process to take more time than simply applying DASrealign, but with the advantage that the reads will also be error corrected. Never the less, just scrubbing reads and feeding them into another assembler is proving to have sufficient value that I decided to release the current pipeline. Best, Gene